


CSC Spring School in Computational Chemistry 2023
siili.rahtiapp.fi/sscc2023
This is a collaborative “notebook” for the CSC Spring School in Computational Chemistry organised 26-28.4.2023 by CSC - IT Center for Science.
 The latest updates on the course schedule can be found in here.
The latest updates on the course schedule can be found in here.
 This is also the place to ask questions about the school content!
This is also the place to ask questions about the school content!
 Hint: HedgeDoc is great for sharing information in this kind of courses, as the code formatting is nice & easy with MarkDown! Just add backticks (
Hint: HedgeDoc is great for sharing information in this kind of courses, as the code formatting is nice & easy with MarkDown! Just add backticks (`) for the code blocks.
Otherwise, it’s like Google docs as it allows simultaneous editing. There’s a section for practice down there ⬇️
💻 How we work
Code of conduct
We strive to follow the Code of Conduct developed by The Carpentries organisation to foster a welcoming environment for everyone. In short:
- Use welcoming and inclusive language
- Be respectful of different viewpoints and experiences
- Gracefully accept constructive criticism
- Focus on what is best for the community
- Show courtesy and respect towards other community members
Using CSC supercomputers (useful links)
- Docs CSC (CSC user guides)
- Connecting to CSC supercomputers
- Puhti web interface
- Linux 101
- Running serial, parallel and interactive batch jobs
- Moving files between local machine and CSC supercomputers using
scpand Puhti web interface
Links and info about hands-on materials (TBA)
Wednesday 26.4
- Slurm advance reservation:
sscc_wed(gpu partition) - Materials available via Puhti web interface
- And https://gitlab.com/hseara/spring_school2023_notebooks
Thursday 27.4
- Lecture slides
- Slurm advance reservations:
sscc_thu_int(interactive partition),sscc_thu_med(medium partition) - QC basic
- QC intermediate
- QC advanced
Friday 28.4
- Materials available via https://gitlab.com/hseara/spring_school2023_notebooks
- VMD slides at https://a3s.fi/sscc/vmd.html
Important info for Friday’s hands-ons!
All notebooks will be run locally on the classroom workstations. This is how you get access to the notebooks and install everything that is needed:
- First, open a terminal (right-click on the desktop and select “Open Terminal”)
- The, run the following commands one by one. Wait for each one to complete before running the following:
git clone https://gitlab.com/hseara/spring_school2023_notebooks.git
cd spring_school2023_notebooks
bash -i dogmi-conda.sh
- Close and reopen the terminal and run the commands:
conda activate csc_spring # ignore the warning
jupyter lab
📅 Agenda
Day 1: Wednesday 26.4.
| Time | Content |
|---|---|
| 8:45 | Registration |
| 9:00 | Welcome and intro |
| 9:15 | Introduction to classical molecular dynamics |
| 10:00 | Break |
| 10:15 | Introduction to classical MD contd. |
| 11:00 | Break |
| 11:15 | Practical info and MD hands-on |
| 12:00 | Lunch break |
| 13:00 | MD hands-on contd. |
| 14:30 | Break |
| 14:45 | MD hands-on contd. |
| 16:15 | Break |
| 16:30 | Poster session Snacks, refreshments |
| 19:00 | Poster session finishes |
Day 2: Thursday 27.4.
| Time | Content |
|---|---|
| 9:00 | Welcome and intro |
| 9:10 | Introduction to quantum chemistry |
| 10:00 | Break |
| 10:15 | Introduction to QC contd. |
| 11:00 | Break |
| 11:15 | Introduction to QC contd. |
| 12:00 | Lunch break |
| 13:00 | QC hands-on |
| 14:30 | Break |
| 14:45 | QC hands-on contd. |
| 16:15 | Transition to sauna lobby |
| 16:30 | Talk, dinner and sauna |
Day 3: Friday 28.4.
| Time | Content |
|---|---|
| 9:00 | Welcome and intro |
| 9:10 | Machine learning in chemistry |
| 10:00 | Break |
| 10:15 | Machine learning in chemistry contd. |
| 11:00 | Break |
| 11:15 | Machine learning in chemistry contd. |
| 12:00 | Lunch break |
| 13:00 | Enhanced sampling methods |
| 14:30 | Break |
| 14:45 | Visualization |
| 16:15 | Closing and goodbyes |
☃️ ICE BREAKER (HedgeDoc -practice)
Ice breakers
Let’s learn how to use this HedgeDoc document by answering an ice breaker question!
Q1: What is your research focused on / what methods have you used in your research/studies so far?
Answers:
- Compuational drug design
- Drug design, protein structure- and ligand based virtual screening, phase, shape screening, homology modeling
- Minerals materials and Petroleum engineering, Gromacs, Lammps, Materials Studio, simulations of water-rock interactions
- Area of calculating decomposition mechanisms of some organic hydrotrioxides present in the atmosphere.
- atmospheric chemistry- mechanism and reaction kinetics
- Molecular dynamics simulation of protein
- MD simulations of biomolecular systems, particularly proteins
- Computational study of HOM formation mechanism
- Chromatographic separation of phytochemicals, new phenomena, first practical studies - then MD (maybe)
- Using Maestro Schrödinger (docking), drug design, virtual screening
- Focus is on transporter proteins: studying transport mechanics and designing prodrugs
- My research is focused on water molecules related to protein kinase inhibitor binding and drug design. So far i have done research with Schrödinger maestero; Docking, Classical MD simulations on desmond
- Protein structure prediction (AlphaFold2, Rosetta)
- Biophysics of proteins, lipids, and lipid bilayer mimetics
- biophysics of lipids and lipid bilayers, MD
- Radiation damage of nuclear materials (TDDFT, Molecular dynamics)
- Study on dynamics and structure of surfactants in bulk water by means of classical MD simulations and modeling techniques for interpretation.
- I am researching identifying 3D structures (lignocellulosic building blocks) from AFM images using ML techniques combined with DFT.
- My research is focused on elucidating the structural dynamics of enzymes at different enzymatic stages by performing molecular dynamics simulations and adaptive sampling. Then, upon the mechanistic understanding of dynamics, I perform virtual screening studies to identify potential orthosteric/allosteric binders. So far, I have used classical MD, aMD, adaptive sampling, and network models, so far.
- CoupledCluster, k-means clustering, DFT, XTB
- Combustion chemistry. Gas phase reaction rate calculations and thermodynamic parameter calculations. I use Orca at the moment with Avogadro
ChatGPT vs. Hector
Q2: What are your expectations for today’s sessions?
Answers:
- Basics, what can be done and what can’t be done.
- Learning the basis of quantum chem
- I know nothing, I want to learn all I can
- Applications for modeling use.
- Getting familiar with the basis of quantum chemistry and gain some experience in the preparation of a system for QM calculations.
- What things i should take into account when designing a QM experiment?
- I would like to learn something new on excited state dynamics. I think that was mentioned on the course description.
- Antti’s opinion on Minnesota functionals.
- How to model reactions
learn the basics of quantum chemistry - I would like to have basic introduction of quantum chemistry and how to use the program CP2K
- I am expecting a good refresher on some familiar topics, maybe from a different perspective, as well as getting to know some more advanced methods.
- I am currently doing TD-DFT calculations, would be nice to know more fundamental theory behind it.
- To learn basics of quantum chemistry and to use the program used in QC because I don’t have any knowledge about the topic
- How quantum computing makes quantum chemistry calculations faster?
- What should we consider to choose the correct basis set and functionals for our systems? is it always a “trial and error” thing?
Q3: Name one interesting/surprising thing you’ve learned during the spring school so far!
Answers:
-
in the lecture we learned that the berendsen thermostat is not a good one but in the mdp files from exercises we used it anyways. ha!
-
(Related to the previous comment, in my nonexpert understanding the berendsen is not good in prod.run, but is robust for equilibrations, and in the exercises it was used there, ha!)
-
Limitations of MD
-
Lecture+hands on: MD doesn’t deal with reactions. Poster: trioxyl radicals, I had no idea about them!
-
That there is a term called experimentalist.
-
that you can calculate so many things easily with QM and that even larger systems are possible in some cases.
-
View system neighboring copies using vmd and measure the distance between them.
-
I learned that I shouldn’t use Euler integration when coding simple classical physics simulations.
-
QC applications and possibilities for my own research
📝 Q & A
Your questions are answered here. We will answer them, and this document will store the answers for you for later use! 
Scroll  to the bottom of the page to submit a question
to the bottom of the page to submit a question
-
I have difficulty pasting my questions into HedgeDoc (here). Do you have some instructions on how to write here?
- A: HedgeDoc can look a bit differend depending on the view you are in. Can you see the three icons on top left corner, next to HedgeDoc text? There’s pencil, this side-by-side symbol, and an eye (see below for a screenshot). In eye view, you can’t edit, you are just viewing. The other two reveal the markdown (MD) version of the page, which you can edit. I find it easiest to edit with the side-by-side view, but it can be a bit slow sometimes. First time opening the page, the small Edit (pencil) button might be on right next to the table of contents. Please note that it might make things faster if you switch to view only mode when not editing!
The small pencil icon for entering edit mode for the first time. You might need to scroll all the way up to see it.

The bigger HedgeDoc toolbar for switching between modes later on.
Hint: You can also apply styling from the toolbar at the top  of the editing area.
of the editing area.
✏️ Add your questions here
Please type your questions ON THE BOTTOM OF THIS SECTION. We will answer them, and organise the document topically.
-
Q0: Have I clicked the edit mode on?
- A: Probably not yet… ↖️
-
Q1: What does brute forcing mean?
- A: Typically it means creating a replica of your system (which means it’s big) and just starting simulation. (dictionary suggests this means throwing lots of resources at the problem instead of doing something clever) I.e. trying to simply repeat what happens in real life. This is very costly in terms of time and computational resources i.e. you will need to wait really long (millions of years). The alternative is to make a clever, much smaller model system that stil captures the physics of your problem and simulate that. And instead of standard molecular dynamics, use some enchanced sampling technique, or e.g. metadynamics to speed up (or parallelize) your simulation.
-
Q2: Why does gromacs need so many different kinds of input files
- A: The main reason is flexibility. Having everything in a single file would not work out in the end and would also result in a lot of redundancy when you are dealing with many systems (you might need to repeat a lot of parameters for each systems separately if there was only one input file).
-
Q3: Does a polarizable force-field yield more accurate results
- A: In theory, yes. But there’s a lot of parameters (poor generalizability) and you might get e.g. over-polarization. Based on experience (Hector) AMOEBA is better than Drude if you need to use a polarizable force field.
- A: Once we have fully automated parametrization processes, polarizable force fields will likely improve significantly. Our expertise on polarizable lipid models is that they haven’t performed very well (benchmarked as a part of the NMRlipids project. (NMRlipids is a great project for lipid force field benchmarking in general). In the meanwhile, the scaled-charge ECC model that Hector quoted a couple of times might be a good compromise.
-
Q4: in CFD simulations we have a dimensionless timestep that is universal for many kinds of systems. Something similar for MD?
- A: Not really, because we almost always have small atoms that require 1-5 fs timestep. The time step is determined by the fastest frequency in the system (those OH, CH bond vibrations) and their robust numerical integration needs a faster evaluation i.e. shorter time step. In some cases if you simulate e.g. only some graphite system with just carbon atoms, or some metal lattice, you could increase it further.
-
Q5: Why not use Berendsen thermostat
- A: Berendsen gives a wrong distribution of individual particle velocities - gaussian (or symmetric) vs Boltzmann.
-
Q6: How do I know which force field to use?
- A: Read papers comparing their behavior to experiments. For lipids, NMRlipids is a good reference. For proteins, there are numerous force field comparison papers, but the results differ between structured and intrinsically disordered proteins.
-
Q7: How do you get around the large number of nuclei and electrons when performing quantum chemical calculations on solid state structures? Are there periodic boundary condition tricks you can perform like in MD?
- A: Yes! Periodic boundary conditions are routinely applied also in quantum chemical simulations of extended systems such as condensed liquid phases or solid crystals.
-
Q8: How to model temperature with quantum mechanics
- A: To model temperature you need time dependence for the nuclei. Thus, the most straightforward method would be ab initio molecular dynamics.
- A2: Another approach is to calculate the frequencies, etc. with Quantum Chemistry, and then use Statistical Thermodynamics to model the temperature-dependent properties. However, this is very difficult when there are time-dependent non-equilibrium phenomena.
-
Q9: In Schrødinger equation there’s so why don’t we just cancel the and get ?
- A: The Schrödinger equation is what we call an “eigenvalue equation”. The Hamiltonian is an operator (a matrix) that operates on the wavefunction (which is a vector). This yields a scalar (or diagonal matrix, one element for each single-particle state), the ground state energy, multiplied by the same wavefunction (a vector). So there’s no “ordinary” multiplication there so no cancellation ;)
-
Q10: So each cell in the Slater determinant is a molecular orbital and those consist of a linear combination of atomic orbitals? Or did I get it wrong?
- A: The Slater determinant itself is a representation for a multi-electron system. It yields an appropriately anti-symmetric wavefunction for a many-electron system. The individual cells in the determinant correspond to single-electron states. These electrons are assumed to be non-interacting.
-
Q11: When does the LCAO model break down
- A: It breaks down when you truncate the linear combination (expansion). If you would use an infinite number of basis functions (numerically not possible) the model would never break down. This is a fundamental property of the wavefunction.
-
Q12: Why is the Hartree approximation a product of single electron wave functions and not a summation
- A: Because the Hamiltonian Operator is a linear operator. If you apply it on a product of functions, the result is a sum of energies. . We know the total energy is a sum of multiple (electron-proton and electron-electron) interactions, and thus a first approximation is to use a product of wvaefunctions describing individual electrons.
-
Q13: Could those coefficients of the basis functions be determined with ML method instead of HF?
- A: Optimizing basis sets can indeed be done with the help of machine learning techniques as well.
-
Q14: How to download the materials?
- A: There are many materials repositories available from where you can download structures for either direct use or to serve as a starting point for creating your own materials. E.g.:
- NoMaD: https://nomad-lab.eu/nomad-lab/
- Materials Project: https://materialsproject.org/
- Open Catalyst Project: https://opencatalystproject.org/
- Computational Materials Repository: https://cmr.fysik.dtu.dk/
- Cambridge Structural Database (CSD): https://www.ccdc.cam.ac.uk/structures/
- A: There are many materials repositories available from where you can download structures for either direct use or to serve as a starting point for creating your own materials. E.g.:
-
Q15: The issue of computation power limitation to get close to an exact solution, can’t GPUs be of any help?
- A: Accelerators like GPUs can speed up calcualtions significantly, yes! But not everything can/should be offloaded to GPUs. GPUs have been designed to be very efficient for solving linear algebra problems similar to those encountered in computer graphics. Quantum chemical calculations involve many routines that are hard to accelerate with GPUs, or would require developers to completely rewrite the code of a QC software. The development is going towards this direction, but many QC software have been developed for decades and the porting (CPU to GPU) effort takes time.
-
Q16: A couple of questions with respect gromacs force fields: 1) How can we define intramolecular vdw for 1-5 and 1-6 (atoms between 4 and 5 bond distances), that are independent from those in [ atomstypes ]? 2) If to gen-pairs is given “yes”, we have 1-4 nonbonded intramolecular interactions, how about 1-5 and above? are they always there the same way as nonbonded intermolecular interactions?
- A: When you define your molecule you define where is the boundary for intra/iter vmd and electrostatics interactions
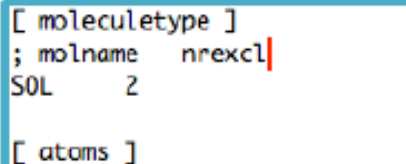
For normal molecules nrexcl is set to 3. This means that intra vdw and electrostatic are those within 3 bonds distances. This means that 1-2, 1-3 are not considered. They are instead encoded using bonds and angles. The boundary 1-4 is encoded in a special way that allows some tuning if needed, e.g., using the [pairs] keyword. But in force field such as CHARMM they are simply normal van der waals and electrostatic interactions on top of the dihedrals.
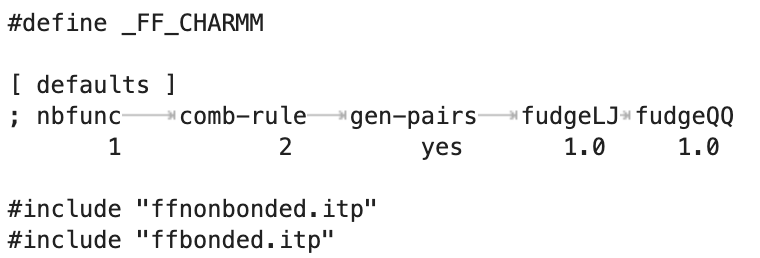
The things get more complex if the general nonbonded interactions of the involved atoms are customized using [ nonbond_params ], as those do not apply to 1-4 pairs. Then for nrexcl=3, pairs beyond 1-4 are simply treated as any other non bonded term.
There is no straight implementation for setting specifically a particular 1-5, 1-6 and so for. But if you really want it can be done by changing their atom types and setting the resulting pairs to particular values using [ nonbond_params ]. b) Yes. Still, if you really would like them to be different you can customize them using [ nonbond_params ] althought it gets very painful.
-
Q17: How to continue learning about machine learning. Any good resources?
- A: A good one is “Elements of statistical learning” by Trevor Hastie, Robert Tibshirani, Jerome H. Friedman. There’s tons of resources available online as well!
- A: Google and others have training material
- A: CSC has various Tutorials and Training material that include data and ML topics.
-
Q18:How do you know when your metadynamics simulation has converged?
- A: (regular) When it seems to sample all over the place. But use the well tempered metadynamics, which indicates convergence.
-
Q19:
- A:
-
Q20:
- A: